Countdown to Compliance: The August 2025 Deadline for NDSRI Testing Looms Over Pharmaceutical Industry
As the pharmaceutical industry approaches the crucial deadline of August 2025, manufacturers of both currently marketed and newly approved drug products are under increasing pressure to evaluate and control the levels of Nitrosamine Drug Substance-Related Impurities (NDSRIs) in their products. The FDA’s recent final guidance on Acceptable Intake (AI) Limits for NDSRIs underscores the critical importance of conducting thorough risk assessments, employing advanced analytical chemistry testing methods, and, when necessary, performing additional mutagenicity testing to ensure compliance and safeguard public health.
The urgency surrounding this issue can be traced back to several high-profile cases that exposed the dangers associated with nitrosamine contamination. Most notably, the Valsartan NDMA contamination in 2018 and the subsequent concerns regarding the formation of carcinogenic and mutagenic NDMA in Zantac brought nitrosamines into the spotlight. These events triggered a global response, prompting regulatory bodies to issue stringent guidelines to prevent such occurrences in the future. As a result, the FDA’s guidance, issued in August 2023, established strict AI limits and set a clear deadline: by August 1, 2025, all drug manufacturers must ensure that the NDSRIs in their products adhere to these limits, or they risk facing severe regulatory consequences, including fines, product recalls, and market access restrictions.

The Role of Nitrosamine Drug Substance-Related Impurities (NDSRIs)
Nitrosamines are a class of chemical compounds that have been identified as potential carcinogens and mutagens, meaning they can cause cancer and genetic mutations. NDSRIs, a specific subset of nitrosamines, are formed when secondary or tertiary amine centers in drug substances react with nitrite sources present in excipients or during the drug manufacturing process. These impurities can form under certain conditions, such as storage conditions, shipping, or as a byproduct of chemical reactions, making their detection and control particularly challenging.
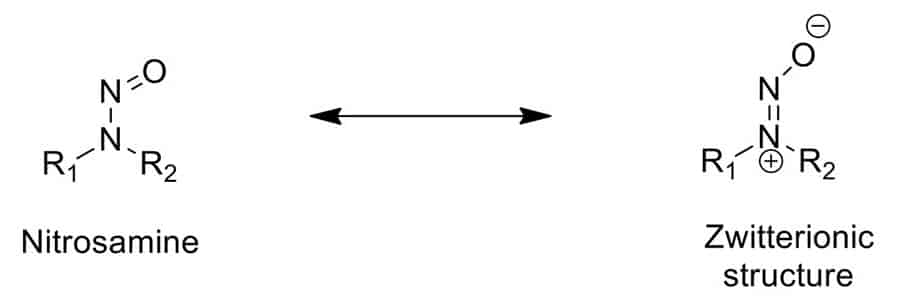
The presence of NDSRIs in pharmaceuticals is of significant concern due to their potential human health risks. Even at trace levels, nitrosamines can pose a serious health threat to patient safety. This concern has prompted the FDA to take a proactive stance, requiring drug manufacturers to conduct comprehensive evaluations of their products to identify and quantify any NDSRIs present.

Analytical Chemistry Testing: A Cornerstone of Compliance
Analytical chemistry plays a pivotal role in ensuring drug safety and compliance with regulatory standards. The ability to accurately detect and quantify NDSRIs at extremely low concentrations—often in parts per billion (ppb) or even parts per trillion (ppt)—is essential for meeting the FDA’s stringent AI limits. Advanced testing techniques such as Liquid Chromatography-Electrospray Ionization-High Resolution Mass Spectrometry (LC-ESI-HRMS) have become indispensable tools for confirmatory testing of NDSRIs.
LC-ESI-HRMS and other similar techniques offer several advantages in the detection of nitrosamines and NDSRIs. These methods provide high sensitivity and specificity, allowing for the precise identification and quantification of impurities, even in complex matrices. The use of high-resolution mass spectrometry is particularly beneficial in distinguishing between closely related compounds, which is critical when analyzing nitrosamines that may have similar structures but different toxicological profiles.
However, simply detecting these impurities is not enough. The data generated from these tests must be thoroughly validated and interpreted within the context of the product’s overall safety profile. This requires not only technical expertise in analytical chemistry but also a deep understanding of the pharmacological and toxicological properties of both the drug and the impurity. Validation of analytical methods is a critical step in ensuring that the testing procedures are reliable and reproducible, providing confidence in the results obtained.

The Importance of Risk Assessment in NDSRI Management
Conducting a comprehensive risk assessment is a critical step in ensuring that a drug product remains safe for consumer use. This process involves evaluating the potential for NDSRIs formation during manufacturing and identifying all possible in-situ as well as other key sources of nitrosating agents and amines. Manufacturers must assess the effectiveness of current mitigation strategies and consider whether any changes to the manufacturing process are necessary to reduce the risk of NDSRI formation.
The FDA’s guidance provides a framework for these risk assessments, encouraging manufacturers to use data from rodent carcinogenicity studies or structure-activity relationships to predict the carcinogenic potency of NDSRIs. In cases where specific data are lacking, the FDA allows for the use of read-across analyses, wherein data from a structurally similar compound is used to estimate the risk posed by the impurity in question. This approach allows for a more informed risk assessment when direct data on a specific NDSRI is not available, but it also requires careful consideration of the structural similarities and differences between compounds.
Risk assessment is not a one-time activity but rather an ongoing process that must be revisited as new information becomes available. For example, changes in raw material suppliers, manufacturing processes, or storage conditions can all impact the potential for NDSRI formation. Regular review and updating of risk assessments are essential to ensure that any emerging risks are identified and addressed promptly.
Mutagenicity Testing: Addressing Uncertain Risks
For NDSRIs with uncertain mutagenic potential, the FDA recommends additional testing, such as an enhanced Ames assay. The Ames assay is a widely used test that assesses the mutagenic risk of a compound by measuring its ability to induce genetic mutations in bacteria. The FDA has adapted the Ames assay to better detect the mutagenic potential of NDSRIs, which often have more complex structures than other nitrosamines traditionally studied.
Conducting this additional mutagenicity testing is not only crucial for regulatory compliance but also for ensuring patient safety. A negative result in a validated enhanced Ames assay can support higher AI limits, which may be necessary in cases where reducing impurity levels is not feasible without compromising the drug’s efficacy. However, a positive result may necessitate further testing or even changes to the drug’s formulation or manufacturing process to reduce the levels of the impurity to an acceptable level.
The process of conducting mutagenicity testing and interpreting the results requires specialized expertise. The design of the testing protocol, selection of appropriate test strains, and analysis of the data all play a critical role in ensuring that the results are reliable and meaningful. Furthermore, the implications of the test results for the overall safety profile of the drug must be carefully considered, taking into account factors such as the intended patient population, dosing regimen, and duration of use.
Meeting the 2025 Deadline: Steps for Compliance
As the August 2025 deadline approaches, pharmaceutical companies must take proactive steps to ensure compliance with the FDA’s guidance on NDSRIs. The following steps outline a comprehensive approach to meeting this deadline:
- Conduct Thorough Risk Assessments: If not already completed, manufacturers should conduct detailed risk assessments to identify drug substances that are potentially susceptible to NDSRI formation. This includes evaluating raw materials, excipients, processing equipment, and packaging materials to identify sources of nitrosating agents and other factors that could contribute to impurity formation.
- Implement Advanced Analytical Testing: Manufacturers should employ advanced analytical techniques, such as LC-ESI-HRMS, to conduct confirmatory testing of their products. These tests should be performed using validated methods that are sensitive enough to detect NDSRIs at trace levels, ensuring that the products comply with the FDA’s AI limits.
- Validate Testing Methods: Validation of the analytical methods used for NDSRI detection is essential to ensure that the results are accurate, reliable, and reproducible. This includes validating the methods for sensitivity, specificity, precision, accuracy, and robustness.
- Perform Mutagenicity Testing as Needed: For NDSRIs with uncertain mutagenic potential, manufacturers should conduct additional testing, such as the enhanced Ames assay, to assess the risk posed by the nitrosamine impurity. The results of this testing should be used to determine whether higher AI limits are justified or whether further mitigation measures are required.
- Develop and Implement Mitigation Strategies: Based on the results of risk assessments and analytical testing, manufacturers should develop and implement strategies to mitigate the risk of NDSRI formation. This may involve an alternate manufacturing processes and reformulating drug products / drug substances, implementing stricter manufacturing controls, or exploring alternative ingredients.
- Collaborate and Communicate with the FDA: Open communication with the FDA throughout the process is essential to ensure that manufacturers are on the right track. Collaboration with suppliers and industry partners can also be critical in navigating the complex regulatory landscape.
- Monitor and Update Risk Assessments Regularly: Risk assessments should be reviewed, discussed and updated regularly to the agencies or in meetings for any changes in manufacturing processes, raw materials, or other factors that could impact the potential for NDSRI formation. This ongoing monitoring is essential to ensure that emerging risks are identified and addressed promptly.
The Ongoing Challenge of NDSRI Control
While the August 2025 deadline represents a significant milestone in the regulatory landscape, the control of NDSRIs is likely to remain an unmet need for human health and going to be the challenge for the pharmaceutical industry. As research continues to advance our understanding of nitrosamines and their potential health risks, manufacturers will need to stay informed about the latest developments and adapt their strategies accordingly.
The FDA’s guidance on NDSRIs is just one part of a broader effort to ensure the safety and efficacy of human drugs. Continued vigilance and proactive management of impurities will be essential to maintain compliance and protect patient safety. This includes not only addressing known impurities like nitrosamines but also being prepared to respond to emerging contaminants as they are identified.
Why Emery Pharma is the Ideal Partner for NDSRI Testing
With the impending FDA guidance and the complexities of NDSRI testing, securing a reliable partner is crucial. Since 2018, Emery Pharma has been at the forefront of developing nitrosamine testing methods using advanced techniques such as LC-MS, GC-MS, HR-MS, and LC-MS/MS. Emery Pharma excels in detecting NDMA, NDEA, and other nitrosamines in human drugs like Valsartan, Zantac, and Metformin. As early pioneers in LC-MS testing for nitrosamines, we ensure robust and validated testing methods. Our state-of-the-art instrumentation, skilled scientific professionals and commitment to quality provide pharmaceutical companies with the confidence to meet stringent regulatory requirements and ensure product safety.
At Emery Pharma, we offer the expertise, advanced capabilities, and dedicated support you need to navigate these regulations effectively. Contact us with confidence today to discover how our tailored solutions can address your nitrosamine testing needs and set you up for a successful journey.
For more information refer to the FDA website: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/updated-information-recommended-acceptable-intake-limits-nitrosamine-drug-substance-related
About the Author
Authored by Dr. Ron Najafi, Emery Pharma’s CEO and Dr. Ryan Cheu, Director of Chemistry.